







| Cellular and Molecular Medicine Research, ISSN 2817-6359 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Cell Mol Med Res and Elmer Press Inc |
| Journal website https://www.thecmmr.org |
Review
Volume 1, Number 2, December 2023, pages 33-43

Stress, Autoimmunity and Mitochondrial Dysfunction in Chronic Obstructive Pulmonary Disease
Alcibey Alvarado
Internal Medicine and Neumology, Clinica de Diagnostico Medico, San Jose, Costa Rica
Manuscript submitted June 5, 2023, accepted August 3, 2023, published online November 7, 2023
Short title: Mitochondrial Dysfunction in COPD
doi: https://doi.org/10.14740/cmmr17e
- Abstract
- Introduction
- Stress
- Autoimmunity and Infection
- Mitochondrial Dysfunction
- Therapeutics
- Conclusions
- References
| Abstract | ▴Top |
Chronic obstructive pulmonary disease (COPD) includes several clinical syndromes, most notably emphysema and chronic bronchitis, respiratory bronchiolitis, asthma and COPD overlap syndrome (ACOS), COPD and obstructive sleep apnea (OSA) overlap syndrome and combination of pulmonary fibrosis and emphysema (CPFE). Many of the current treatments fail to attenuate the severity and progression of the disease, so a better understanding of the pathogenesis of COPD is required to develop treatments that modify it. Various types of stress are now recognized as predisposing factors in the pathogenesis of COPD. There is increased evidence of the presence of autoantibodies in COPD. Oxidative stress, for example, can lead to increased levels of reactive carbonyls in the lung, which could result in the formation of carbonyl adducts in highly immunogenic and potentially destructive on “self” proteins. This establishes a correlation between autoimmunity and COPD. Recent studies show that mitochondria are involved in the innate immune response signals, which play important roles in the activation of airway inflammation induced by cigarette smoke, lung inflammation and tissue remodeling. The connection between these three elements in the pathogenesis of COPD is discussed here. Finally some therapeutic alternatives that can impact these elements are reviewed.
Keywords: Chronic obstructive pulmonary disease; Stress; Autoimmunity; Mitochondrial dysfunction
| Introduction | ▴Top |
Chronic obstructive pulmonary disease (COPD) is a global public health problem. It is projected that in 2020 it will be the third leading cause of death worldwide, and the fifth cause of years lost due to disability along with years lost due to premature death (DALYs) [1]. This condition affects 10% of the world’s population above the age of 45, and not only 15% of total smokers, but also 50% of heavy smokers [2, 3]. The definition states that COPD is due to an increased chronic inflammatory response [4]. COPD is characterized not only by inflammation but also by remodeling of the small airways and destruction of the lung parenchyma (emphysema) [5]. The major pathogen that causes inflammation appears to be stress. For example, oxidative and carbonyl stress occur from exposure to cigarette smoke and/or biomass fuels [6]. Oxidative stress is due to exposure to reactive oxygen species (ROS), which in turn damages the surrounding tissue forming highly reactive organic molecules (reactive carbonyls) that can modify host proteins, markedly immunogenic, generating autoimmunity in COPD [7]. ROS may be exogenous or endogenous, as mitochondria are an important source of ROS in many mammalian cells [8]. In turn, mitochondria are susceptible to oxidative damage, which causes disturbance of mitochondrial function and are also involved centrally in the control of signals that regulate innate immunity and adaptive immunity [9, 10]. Multiple studies have defined that mitochondrial function and its molecules may contribute to the pathogenesis of COPD, establishing a possible connection among the various types of stress, autoimmunity and mitochondrial dysfunction in the entity [11]. The objective of this work was to investigate and dissect the interrelationship among these three pathogenic elements and to update some therapeutic tools that are being implemented at sub-cellular level in the treatment of COPD.
| Stress | ▴Top |
Cigarette smoke (CS) and other pollutants/biomass fuels are the initial noxa that stimulates epithelial cells and macrophages to release chemotactic factors that attract T cells, neutrophils and fibroblasts [12, 13]. T cells are CD8+ (suppressor/cytotoxic subtype Th1/Tc1 and Th17) and release granzymes, perforines and tumor necrosis factor alpha (TNF-α) which cause apoptosis and direct damage to lung parenchyma, and also release IL-4 and IL-13 that induce hypersecretion of mucus in the airways [14, 15]. Macrophages/neutrophils also play an important role in releasing proinflammatory cytokines such as IL-8 and TNF-α and proteases, resulting in inflammation and direct destruction of the lung parenchyma [16]. The CS contains in each inhalation around 1017 ROS which initiate the inflammatory response in macrophages and epithelial cells.
Oxidative stress has a pivotal role in the pathogenesis of COPD (Fig. 1). Sources of exogenous ROS include CS, oxidant gases, ultrafine matter particles, environmental pollution nanoparticles, exhausted vapors, and biomass fuels for cooking and heating households (particularly in third world countries) [17, 18]. Endogenous oxidants result mainly from mitochondrial respiration and inflammatory response to viruses and bacteria (which are common causes of acute exacerbations in COPD). Inflammatory stress is mediated by IL-1, TNF-α and interferon-γ generating endogenous ROS. Other sources of intracellular ROS are the enzyme nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, xanthine oxidase (XO) and hem-peroxidases, all of which are increased in bronchoalveolar lavage (BAL) in patients with COPD. These enzymes catalyze the production of free radicals [19]. The most destructive oxidant molecules are H2O2 (which act as a substrate to generate additional oxidant molecules), ethane and isoprostanes (produced by direct oxidation of arachidonic acid).
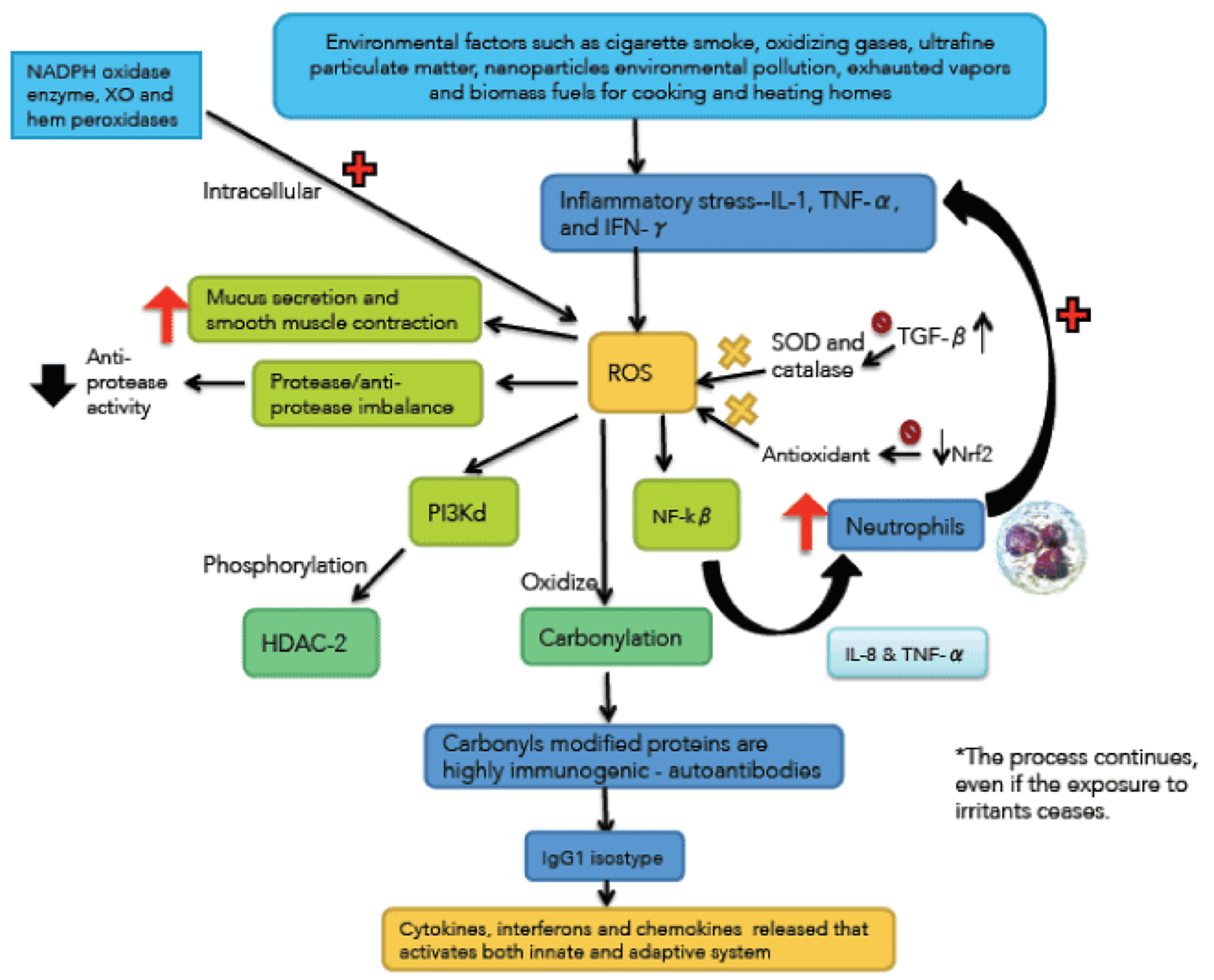 Click for large image | Figure 1. Immunological and inflammatory profile in COPD. Environmental factors produce different types of stress that generates small airways lesion and emphysema, activation of inflammatory cells and autoimmunity. Neutrophils generate more inflammatory stress and autoimmunity actives immune responses that amplify and perpetuate the inflammatory process. The negative sign means repression and positive sign activation. Straight arrows mean increase. Curves arrows mean activation and/or expression. |
An atom contains a nucleus, around which electrons usually move in pairs. A free radical is an atom or molecule that contains one or more unpaired electrons, making them more reactive than the corresponding non-radical [20]. Some of the most powerful oxidants are free radicals. Molecular oxygen is vital for energy that is essential for life [21]. Hypoxia-inducible factor-1 (HIF-1) mediates the adaptive response experienced by cells that survive oxygen deprivation. One pathway promoted by HIF-1 for cell survival under hypoxic conditions is the transition from oxidative to glycolytic metabolism. Under aerobic conditions, electrons are transferred from NADH, and flavin adenine dinucleotide (FADH2) to mitochondrial complex I and II, then to complex III and finally to IV, where electrons react with oxygen to form water [22]. Under conditions of hypoxia (as in COPD, particularly exacerbated), the release of electrons is increased prior to transfer to the IV complex, resulting in the production of superoxide anion, which is then converted to H2O2 and other ROS. The human body therefore generates superoxide anion by adding a single electron to oxygen [23].
O2 + e- --- O2•-
Exposure to high energy radiation causes rupture of one of the covalent bonds of the water generating the hydroxyl radical, OH•.
H-O-H --- H• + OH•
As there is always some degree of environmental radiation, the hydroxyl radicals are always produced in the body. They attack proteins, lipids (damaging cell membranes) and DNA inside the cells producing a chain reaction, which can spread for years favoring neoplasms. Some free radicals are helpful, to a certain extent, like nitric oxide (NO•), which is produced in almost all tissues. In the walls of blood vessels helps to control blood pressure, but nitric oxide and superoxide anion form the peroxynitrite (O2•- + NO• --- ONOO•) that nitrosylates protein amino acids and enzymes, inactivating them. This is known as nitrative stress, which in turn produces more endogenous hydroxyl radical [24]. Therefore, ROS and reactive nitrogen species (RNS) are continuously generated at the cellular level by mitochondrial metabolism and by inflammatory cells in the case of hypoxic diseases such as COPD.
Free radicals, unstable by default, initiate an oxidative process, and a number of adverse consequences at the cellular level. For example, oxidants activate nuclear factor kappa-beta (NF-kβ) increasing IL-8 and TNF-α synthesis which recruits more neutrophils and perpetuating inflammatory stress. Oxidative stress also activates phosphoinositide 3-kinase-delta (PI3k-ð), which phosphorylates histone deacetylase 2 (HDC-2) (a key anti-inflammatory enzyme), and inactive it, subsequently suffering ubiquitination and proteosomic degradation [25]. There is an interaction between pathological mechanisms and oxidants that contribute to the imbalance between proteases/antiproteases, by reducing antiprotease activity. This is the case of myeloperoxidase (MPO) derived from neutrophils, which produces the highly destructive hypochlorous acid, and in turn, it inactivates alpha1-antitrypsin (α1-AT). Oxidants also mediate plasma exudation, increase mucus secretion and smooth muscle contraction [24].
In non-pathological conditions, lungs constant exposure to exogenous and endogenous sources of oxidative stress, contributes to the development of potent antioxidant strategies. For example, up to 20% of reduced glutathione (GSH) is located within the mitochondria, disposed to neutralize endogenous ROS (byproduct of metabolism). Also, the lung environment-exposed surface contains antioxidants, such as ascorbic acid (vitamin C), α-tocopherol (vitamin E) and bilirubin. Larger molecules, such as albumin and mucin, have exposed sulfhydryl groups that act as antioxidants as well. Several studies have described an association between altered lung function and low levels of antioxidants in the lung, suggesting that oxidative stress occurs due to increased exposure to ROS (exogenous and endogenous) and reduced antioxidant capacity (overwhelmed or genetically altered) [7, 26].
Transforming growth factor-beta (TGF-β) expression is increased in EPOC (produced by epithelial cells in response to oxidative stress), and it inhibits the expression of antioxidant enzymes catalase and superoxide dismutase 2 (SOD 2). Both are critical for neutralizing mitochondrial ROS and are under the control of the forkhead box class O (FOXO3) transcription factor, whose deficiency has been associated with COPD [27]. About 200 cellular antioxidant and detoxification enzymes are under the control of nuclear erythroid 2-related factor (Nrf2) [28]. COPD patients have reduced expression and activity in Nrf2. Upregulation or restoration of Nrf2 activity may be beneficial in COPD [29]. Endoplasmic reticulum (ER) oxidative stress can induce mitochondrial apoptosis and cell death. Normally a cross-communication between ER and mitochondria prevents apoptosis of epithelial cells [30].
| Autoimmunity and Infection | ▴Top |
An essential feature of COPD is that the inflammatory process and stress continue after stopping exposure to bronchial irritants [31]. Autoimmunity and recurrent infection are likely to be responsible for this behavior. Autoimmunity has been questioned as a perpetuating mechanism of the inflammatory process and some recent works with little statistical power do not find a pathogenic role [32]. Other papers found significant associations between COPD and autoimmune diseases. For example, women with primary Sjögren syndrome (pSS) are at greater risk of developing COPD than women without pSS [33], and middle-aged women with unexplained chronic cough and lymphocytic airway inflammation, lymphopenia and autoantibodies, are eight times more likely to have autoimmune diseases than controls. Some will develop non-smoked COPD [34]. Although it is not clear how far the immune response is a protective or destructive epiphenomenon, the evidence actually points to an etiopathogenic role of the phenomenon in COPD.
ROS induce the formation of carbonyl adducts in certain host proteins (Fig. 2). The aldol reaction in chemistry refers to the formation of carbon=carbon bonds (aldehydes and ketones), in this case between ROS and proteins. What happens is a covalent bond between ROS and certain amino acid residues, for example, with the sulfhydryl groups of cysteine, ε-amino of lysine, and imidazole of histidine. In this pathway protein adduction is produced by oxidized lipids containing carbonyls derived from polyunsaturated fats (PUFAs) in cell membranes (lipid peroxidation). There is also another direct pathway oxidation in amino acid residues such as arginine, proline and threonine [35]. One characteristic of protein carbonylation is that it is a non-enzymatic, irreversible process leading to various types of protein dysfunction. Moderately carbonylated proteins are degraded by the cellular proteasomal system, but markedly carbonylated proteins tend to form high molecular weight aggregates that are resistant to degradation and accumulate as damaged and unfolded proteins, which inhibits the proteosomal activity. In other words, they are resistant to proteolysis [36-38]. In fact, quantifying carbonylated proteins by various essays is the most widely used method to measure protein oxidation [39].
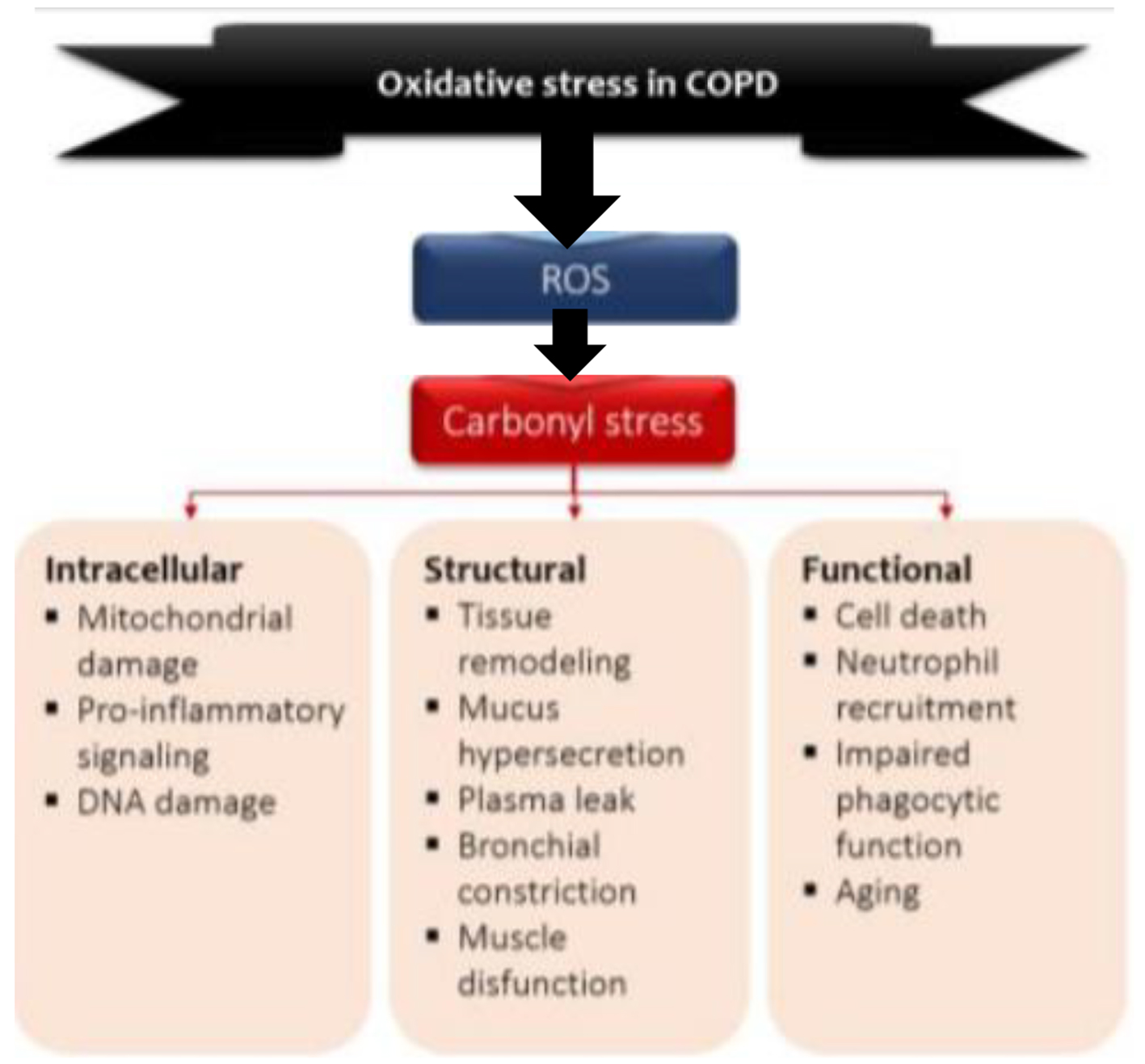 Click for large image | Figure 2. Model of the role of oxidative stress in COPD. Environmental and endogenous sources of reactive oxygen species (ROS) cause lipid peroxidation and the oxidation of proteins and carbohydrates, leading to carbonyl stress. Carbonyl stress in turn produces detrimental intracellular, structural and functional process. O2•: superoxide radical; OH•: hydroxyl radical; ONOO•: peroxylnitite; H2O2: hydrogen peroxide. |
Carbonylated proteins are highly immunogenic and lead to the formation of neo-antigens in the form of carbonylated self-protein, which leads to the generation of autoantibodies that are elevated not only in serum of patients with COPD but also in pulmonary epithelium and endothelium of the pulmonary vessels [40]. The identity of endothelial antigens is yet to be solved, but this may be a fertile field of research into the pathogenesis and treatment of pulmonary hypertension in COPD. Several carbonylated proteins are converted into antigens. There are described antibodies to elastin, human serum albumin (HSA) and from pulmonary parenchyma, malonyldialdheyde protein, and acrolein-modified protein [35]. Circulating and in-lung antibodies are from the IgG1 isotype directed against carbonyl epitopes and could be the result of an aberrant immune response directed against putative antigens for which immune tolerance has been lost or never acquired. These autoantibodies bind complement (C3) and may contribute to the production of emphysema [41]. The presence of these reactive carbonyls and autoantibodies correlates inversely with FEV1 and the severity of the disease [42].
Carbonylated proteins are recognized by innate immunity through pattern recognition receptors (PRRs), expressed in cells that identify antigens, such as dendritic cells and macrophages [43]. In these cells, they are processed and re-expressed in association with the major histocompatibility complex-2 (HLA-2). This promotes the activation of the acquired immune response by attracting Th1 and Th17 cells to lung parenchyma and dendritic cells in small airways. Dendritic cells (DCs) are inflammatory professional antigen presenting cells (APCs), which play a central role in orchestrating immune responses and are essential in the connection of the innate and adaptive immune response and in sieving the environment for the presence of “dangerous signals” from epithelial surfaces [44]. Besides producing neo-antigens, the immune response also promotes the influx of the immune cells needed for recognize and process them. This stimulus causes the release of CCL2 and CCL20 (MIP3ð), which recruit DCs, monocytes and lymphocytes, amplifying the immune response. IL-17 and IL-18 levels are increased. These interleukins activate and mature B cells, and also promote autoimmunity [45-50]. The genetic basis of emphysema is consistent with a multifactorial heritable trait that is common to many autoimmune genetic defects [51]. Although CS can cause recruitment and activation of inflammatory cells in the lung, not all smokers develop the clinical picture of lung disease. The exact prevalence of emphysema is unknown at present, and to date, the known gene that increases the risk of early emphysema is the mutation in the α-1AT protein, encoded by SERPINE 1 gene, but many other loci have been considered [52-54]. Even among carriers of different forms of α-1AT protein, there is great phenotypic variability, suggesting that genes and environmental modification together exert a combined effect.
While CS is the major risk factor for COPD, respiratory infections may play a role in the development and progression of the disease, and are also the leading cause of acute exacerbations [55]. CS and respiratory infections carry different pathways to the activation of multiple PRRs. In the case of CS, ROS or molecules released by epithelial damage stimulate the receptors in the airway cells, which initiate and perpetuate the inflammatory process. These molecules are known as damage-associated molecular patterns (DAMPs). In the case of viral or bacterial infection, pathogen-associated molecular patterns (PAMPs) are released [56]. The PRRs are germ-line encoded, evolutionarily conserved molecules and consist of Toll-like receptors (TLRs), NOD-like receptors (NLRs), RIG-I-like receptors (RLRs), C-type lectin (CLRs) and cytosolic DNA sensors [57]. These receptors sense viral and bacterial PAMPs and DAMPs produced by infectious or non-infectious injury of airway epithelium and/or lung parenchyma. NLRP1-3 generates inflammasomes, which are aggregates of multiple proteins that mediate inflammation. These PRR-expressing cells produce cytokines, interferons and chemokines that recruit macrophages, neutrophils and activate epithelial cells, the innate immune response. DCs, stimulated by ligands in their PRRs and associated with HLA-2, initiate signals that attract T cells, the adaptive immune response.
Patients with COPD have increased colonization by H. influenza, S. pneumoniae, P. aeruoginosa and M. catharrhalis, and this contributes to chronic inflammation and airway dysfunction. Infection with these and other pathogens (including viruses) is a major cause of acute exacerbations [55]. Increased susceptibility of COPD patients to these infections appears to be related to a dysfunctional innate immune system and altered mucociliary clearance.
It is important to emphasize the role of PRRs in COPD, since they mediate infectious and autoimmune responses, the two events involved in the persistence of the inflammatory process. Process becomes autonomous and does not disappear, even after exposure to irritants ceases.
| Mitochondrial Dysfunction | ▴Top |
The human lung is composed of approximately 40 cell types, regionally and spatially located through the organ, and contains several levels of mitochondria [58]. The mitochondrial genome is inherited through the maternal germ line and has been inherited from aerobic prokaryotes bacteria more than a billion years ago and retains many of the morphological and biochemical characteristics of their bacterial ancestors. They are an icon of convoluted double-crested structures that are present in all cells of the body and have their own genome, transcriptome and proteome [59]. According to the endosymbiotic theory, the mitochondria are descendants of old bacteria that entered symbiotic relation with the cells of the host [60]. Conceptualized as “cell batteries” (and consistent with this analogy subject to change), mitochondria are complex cellular organelles assembled from proteins encoded by two distinct genomes: nuclear chromosomal DNA and the mitochondrial genome, i.e., mitochondrial DNA (mtDNA). Despite the small size of the mtDNA (16.5 kb in humans), it encodes 13 potential oxidative phosphorylation subunits (OXPHOS) which interact with more than 70 units encoded by nuclear DNA: their concerted action is necessary to produce ATP, which is required for all active cellular processes [61]. Historically, the major role of mitochondria has been to catalyze the oxidation of metabolites for the production of ATP, via OXPHOS. OXPHOS involves transferring high energy electrons derived from NADH and FADH2 to the mitochondrial complex I and II, then to complex III and finally to complex IV, where they react with oxygen to form water. During this process, the protons are pumped through the inner mitochondrial membrane resulting in a proton-motive force which is used by the FOF1 ATP synthase [22, 62]. Mitochondria continually oxidize fatty acids and consume end products of glucose, glutamine and amino acids for the production of ATP. Lung mitochondria preferably use glucose-derived substrates such as pyruvate for the production of oxidative energy. It also has its own electron transport chain (ETC), complex IV cytochrome c-oxidase (COX subunit IV-2) sensitive to oxygen and doubly active (binding oxygen) compared to COX from other tissues [63].
Additional critical functions of mitochondria such as regulation, proliferation, differentiation, cell death, redox and calcium homeostasis have been revealed during the last two or three decades. Recently, research has pointed to mitochondria as controlling the immune responses and determinants of immune cell phenotypes and their functions, including CD4+ T cell differentiation and CD8+ memory T cell formation [64]. Mitochondrial aerobic glycolysis is required for effective activation of T cells through the generation of mtROS, which are necessary for optimal activity of nuclear factor of activated T cell (NF-AT) and proximal T-cell receptor-mediated signaling [65].
While elevated levels of ROS produce tissue damage and promote aging, low ROS levels enhance defense mechanisms by inducing adaptive immune response (mitochondrial hormesis or mithormesis) [66]. Other fundamental concepts of the activity and function of mitochondria are mitochondrial biogenesis and mitophagy. Both refer to the phenomena of fusion and fission of the mitochondria. In fusion two mitochondria bind to generate another mitochondria while in fission, a mitochondria is divided to generate two. This phenomenon allows the cells to replace metabolically dysfunctional mitochondria with fresh, undamaged mitochondria and allows the cells a healthy pool of mitochondria, since they do not form of “novo”, but the biogenesis results in growth and division of pre-existing mitochondria [67]. Mitophagy is a selective encapsulation by double membrane autophagosomes that is delivered to lysosome for degradation and removes damaged mitochondria that generate excessive amounts of ROS that can damage the mitochondria or other cellular components themselves [68, 69]. Metabolically active cells (e.g., type II pneumocytes) also known as alveolar epithelial cells (AECs) have developed robust mitochondrial quality control programs.
Within the mitochondrial proteome are key proteins such as mitochondrial antiviral signaling protein (MAVS), which is the first protein located in the mitochondria involved in innate immune response and inflammatory response [70]. MAVS interacts with many molecules that play a role in apoptosis, mitochondrial dynamics, autophagy and proteosomal degradation. NOD-like receptor (NLRX1) is a member of the NLR family of PRRs that has a unique N-terminal domain (which explains the letter X1 in the acronym) and is the first, and only so far, of the PRRs located in mitochondria and therefore, it establishes a connection between mitochondrial functions and innate immunity [71]. Alveolar macrophages are essential for lung defense of the host through its ability to survive and regulate the innate and adaptive immune response. They are believed to be maintained in a relatively “quiescent state” with active suppression of inflammatory response to harmless antigens to prevent collateral damage to lung tissue [72, 73]. Apparently, and primarily by experimental investigation in the murine model of COPD, this inhibition of inflammation and remodeling is performed by NLRX1 regulating MAVS [11].
The presence of two genomes in human cells - the nuclear genome and the tiny mtDNA - is a curiosity of evolution. Altered mtDNA diseases (more than 400 mutations or deletions in the 16,569-bas-pair mitochondrial chromosomes that contain only 32 genes), are heterogeneous disorders with well-known genetic causes [74]. The possibility of mitochondrial replacement in certain individualized cases is a viable therapeutic option. In fact, on December 15, 2016, the Human Fertilization and Embryology Authority in the United Kingdom approved the use - in certain specific cases - of an in vitro fertilization (IVF) technique involving donation of mitochondria [75]. The discovery of pathogenic defects of mtDNA occurred in the 1980s, but since then much research has revealed a number of common diseases, in which mitochondrial dysfunction as a pathogenic and/or perpetuating mechanism of the process underlies. COPD does not escape this fact [76].
In COPD, CS exposure reduces mitochondrial OXPHOS in airway smooth muscle cells, quadriceps, and external intercostal muscles, compromising oxidative function [77]. Alteration of mitochondrial biogenesis may be associated with a significant reduction in body mass index and lower extremity muscle mass, a common phenomenon in COPD [78]. Patients with COPD have increased mitochondrial fission and mitophagy, and pathogenic defects and/or loss of mtDNA have been associated with COPD [79]. Three independent studies have shown that NLRX1 protein expression is suppressed by CS in patients with COPD and this correlates with airflow obstruction. Basically, the idea is that CS alters the state of quiescent homeostasis of the alveolar macrophage by inhibiting the expression and activity of NLRX1, inducing NLRX1/MAVS-mediated mitochondrial dysregulation. CS on inactivation of NLRX1 increases inflammatory activation (IL-18) favoring emphysematous destruction, mitochondrial ROS and proteases matrix metalloproteinases (MMPs) [11, 65].
Mitochondria regulate four mechanisms of cell death: extrinsic apoptosis, intrinsic apoptosis, necrosis/necroptosis and pyroptosis. All have been documented in lung diseases, including COPD. The disturbance in mitochondrial function induced by CS decreases the ability of the mitochondria to generate ATP, producing a switch from apoptosis to necrosis. ATP from damaged or dying cells acts as DAMPs, much like mtDNA, perpetuating the inflammatory cascade. Increased levels of ATP have been found in the BAL of patients with COPD [80, 81].
In this way, the three mechanisms involved, namely, various types of stress, autoimmunity and infection and mitochondrial dysfunction sometimes act in parallel, sometimes in series and often concert, generating, amplifying and perpetuating the inflammatory phenomenon of the entity (Fig. 3).
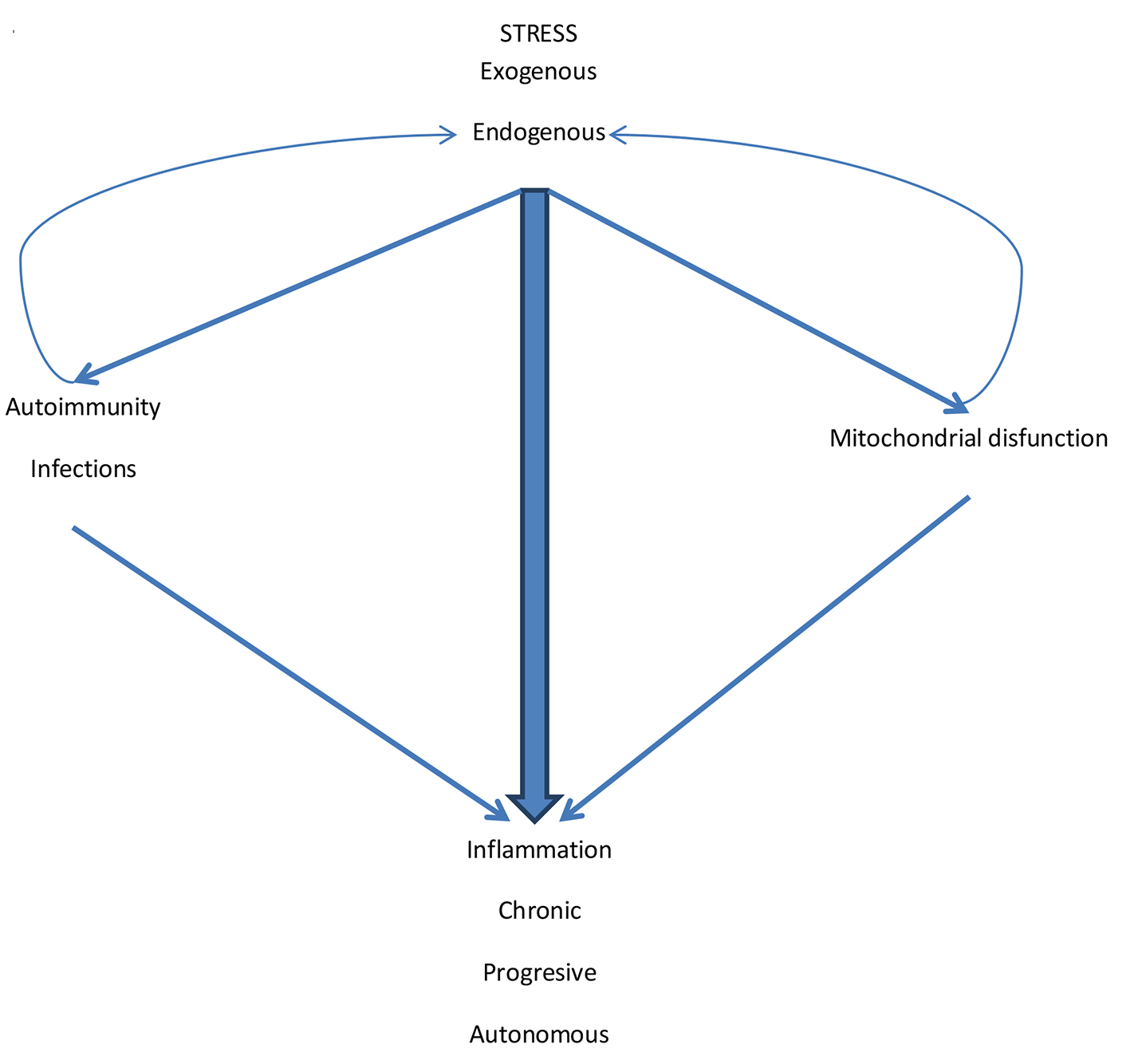 Click for large image | Figure 3. The pathogenic mechanisms of inflammation in COPD. Exogenous stress initiates the inflammatory process. Endogenous stress perpetuates it. Although the external stimulus (CS and other pollutants/biomass fuels) is suspended, endogenous stress and cyclic infections facilitate autoimmunity. Also the various types of stress generate mitochondrial dysfunction. Autoimmunity and mitochondrial dysfunction generate more inflammation but also increase endogenous stress by amplifying the inflammatory process that becomes chronic, progressive and autonomous. |
| Therapeutics | ▴Top |
Existing pharmacological treatments do not reduce the progression of COPD. Bronchodilators (which are the pharmacological basis of therapeutics) only provide symptomatic relief [82]. Glucocorticoids have no effect on disease progression or mortality. A slight reduction in exacerbations has been reported, but this effect has recently been questioned [83]. Medical and pharmacological investigations have investigated alternatives that try to impact the inflammatory process, and the mechanisms that perpetuate it. Some recent options are discussed below.
Antioxidant alternatives
Small molecule thiol antioxidants such as N-acetyl cysteine (NAC) have direct and indirect properties in COPD. NAC free-thiol group is capable of interacting with the electrophilic groups of ROS. Indirectly, NAC is a precursor to GHS, a neutralizer of ROS. NAC may serve as a protective factor against exogenous and endogenous ROS [84]. The BRONCUS study failed to show any effect of orally administered NAC (in conventional doses), in reducing progression of the disease or frequency of exacerbations. Although there was an apparent benefit in patients with COPD treated with NAC but without inhaled steroids [85]. The results may be due to failure of NAC to operate in the subcellular compartment or by insufficient dose of the drug or frequency. The HIACE study (1,200 mg/day oral NAC for 1 year) showed a significant improvement in the small airways function and reduced frequency of exacerbations [86]. An Israeli study found beneficial effects of NAC on trapped gas [87]. However, no effect on quality of life or symptoms has been demonstrated in patients receiving NAC [88]. Global Initiative for Chronic Obstructive Lung Disease (GOLD) does not recommend regular use of these drugs, but recognizes that with or without inhaled steroids, the use of high doses of NAC significantly reduces exacerbations, particularly in patients with stage 2 spirometry [4]. New activators of Nrf2 prevent oxidative stress induced by autoimmunity [7]. Nrf2 regulates almost all antioxidants and phase II cytoprotective genes [89]. Within the activators of Nrf2 activators are the sulforophanes, phytodrugs present in broccoli and other cruciform vegetables, but are not always very potent. New Nrf2 activators are significantly more potent than sulforophanes, and despite their great potential in COPD, they present possible concerning safety issues [90, 91].
Inhibition of oxidative enzymes could be a promising approach. Xanthine oxidoreductase (XO) has been implicated in the development of tissue damage due to oxidative stress in cardiovascular and respiratory diseases. The level of XO is four times higher in the sputum and BAL of patients with COPD than the healthy controls. The use of XO inhibitors such as allopurinol and febuxostat may offer some benefit [92]. Celestrol inhibits the four isoforms of NOX (NADP oxidase), and has a potential use in inflammatory diseases [93]. AZI (an MPO 2-thioxanthine inhibitor) appears to downregulate the inflammatory response induced by to CS, with the advantage that the inhibition is irreversible (i.e., suicide inhibitor). This could be of benefit in COPD, since most of the current inhibitors are short-acting or reversible [94].
Within the “radical scavenger”, edaravone is a powerful free radical neutralizer, reducing carbonyl stress and lipid peroxidation, therefore have a future potential in COPD. Lazaroids are non-glucocorticoid analogs of methylprednisolone that penetrate hydrophobic membranes regions and inhibit lipid peroxidation. Its protective effects have been described in animal models of pulmonary injury; both require studies for their potential use in humans with COPD. Spin traps are compounds that can stabilize free radicals to form stable end products. They have been widely used in vitro and their therapeutic effects have also been investigated in vivo in animal models, showing some benefit [89].
Enzymatic antioxidants are small molecules with catalytic properties that can mimic the activity of antioxidant enzymes. Those who resemble SOD (i.e., SOD mimetics) are the macrocyclic ligands based on manganese, manganese-metalloporphyrins and salens (which also block nitrative stress). The development of SOD-like molecules seems to be a rational therapeutic intervention in emphysema [95]. Ebselen, an organic selenium-based compound, neutralizes oxidative and nitrative stress by resembling glutathione peroxidase activity. Inhibition of inducible nitric oxide-synthase (iNOS) by various chemical complexes may provide a strategy in COPD management. Inhibitors of iNOS may be helpful in the treatment of pulmonary inflammatory disorders [96].
Thioredoxin (Trx) and redox effector factor-1 (Ref-1) belong to the oxidoreductase family of redox sensors. Trx inhibition resulted in diminished neutrophil influx and TNF-α production in animal models. Activation of Trx can attenuate oxidative stress. The antioxidant action of ambroxol at physiological concentrations is at least partially mediated by TrxR and/or the Trx system. Trx effects on respiratory diseases remain to be investigated [97].
Immunological modulation
It is possible that in the future, immunological modulation will be beneficial as treatment for this entity. The development of selective carbonyl blockers may lead to diagnostic and therapeutic tools and some monoclonal antibodies are under development [39, 41, 46]. For example antibodies directed against IL-17 could be a promising treatment [50].
Inhibitors of the inflammatory pathway IL-1β (IL-1Ra/anakinra, IL-1 Trap/rilanocept) or using neutralizing antibodies against IL-18 could limit destruction and remodeling in COPD [98, 99].
The administration of antioxidants is a viable alternative from the perspective of preventing or ameliorating autoimmune diseases, particularly by increasing evidence of oxidative damage generating autoimmunity. However, this should be evaluated in light of the disappointing results obtained with antioxidants in cardiovascular diseases [100].
Indeed this therapeutic approach to COPD is found in childhood.
Mitochondria as a therapeutic objective
Mitochondria offer research objectives not only for diagnosis but for treatment of lung diseases. Transfer of mitochondria from bone marrow-derived mesenchymal stem cells to injured alveolar cells could be beneficial in COPD [65]. Antioxidants directed against mtROS may be more effective than extracellular antioxidants like NAC. Attempts to block mtROS should be careful since low doses of mtROS are cytoprotective (mithormesis). OXPHOS could be an objective to intervene as well as to create strategies to reduce mitophagy [63, 79]. Several proteins have been implicated in the communication between the ER and the mitochondria and that contact occurs prior to the phenomenon of fission. Contact is important for the transmission of ER calcium signals to mitochondria. Therefore, designing strategies that refine and perpetuate transmission may improve the function and survival of mitochondria [101]. As in immunomodulation, the attempt to modulate mitochondrial function as a therapeutic target is only initiated.
| Conclusions | ▴Top |
1) The inflammatory process in COPD is chronic and autonomous.
2) The inflammation persists even if the initial stimulus has been discontinued.
3) This explains the deterioration in lung function and the progression of the entity.
4) This fact is due to the persistence of various types of stress of endogenous origin, to the autoimmunity generated by them, to the cyclic infection of these patients and to mitochondrial dysfunction.
5) These elements act in concert to give autonomy, persistence and amplification to the inflammatory process.
6) The current treatment of COPD produces symptomatic improvement and health-related quality of life, but it improves little, if at all, the inflammatory phenomenon.
7) More and better primary and clinical research is needed to find new and powerful drugs that impact sub-cellular compartments and metabolic pathways for more effective therapy in this entity. There is no single magic bullet to combat all the pathogenic mechanisms, so combined therapy may be more effective in COPD.
8) Pharmacology and medical science should integrate research to design these molecules and demonstrate their efficacy and safety in powerful, controlled, and randomized clinical trials.
Author Contributions
This work was only carried out by the author. AA contributed in the planning, data collection, data analysis, writing and critical review. AA read and approved the final manuscript.
Source of Support
None.
Conflict of Interest
None.
| References | ▴Top |
- Lopez AD, Murray CC. The global burden of disease, 1990-2020. Nat Med. 1998;4(11):1241-1243.
doi pubmed - Lundback B, Lindberg A, Lindstrom M, Ronmark E, Jonsson AC, Jonsson E, Larsson LG, et al. Not 15 but 50% of smokers develop COPD? - Report from the obstructive lung disease in northern Sweden studies. Respir Med. 2003;97(2):115-122.
doi pubmed - Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360(23):2445-2454.
doi pubmed - Global strategy for the diagnosis, management and prevention of COPD. Global Initiative for Chronic Obstructive Lung Disease (GOLD). Available: http:www.goldcopd.org/. (Accessed 23 April 2017).
- Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645-2653.
doi pubmed - Salvi S, Barnes PJ. Is exposure to biomass smoke the biggest risk factor for COPD globally? Chest. 2010;138(1):3-6.
doi pubmed - Kirkham PA, Barnes PJ. Oxidative stress in COPD. Chest. 2013;144(1):266-273.
doi pubmed - Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1-13.
doi pubmed pmc - West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011;11(6):389-402.
doi pubmed pmc - Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. 2015;42(3):406-417.
doi pubmed pmc - Kang MJ, Yoon CM, Kim BH, Lee CM, Zhou Y, Sauler M, Homer R, et al. Suppression of NLRX1 in chronic obstructive pulmonary disease. J Clin Invest. 2015;125(6):2458-2462.
doi pubmed pmc - Hunninghake GM, Cho MH, Tesfaigzi Y, Soto-Quiros ME, Avila L, Lasky-Su J, Stidley C, et al. MMP12, lung function, and COPD in high-risk populations. N Engl J Med. 2009;361(27):2599-2608.
doi pubmed pmc - Wilk JB, Chen TH, Gottlieb DJ, Walter RE, Nagle MW, Brandler BJ, Myers RH, et al. A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet. 2009;5(3):e1000429.
doi pubmed pmc - Saetta M, Turato G, Maestrelli P, Mapp CE, Fabbri LM. Cellular and structural bases of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;163(6):1304-1309.
doi pubmed - Barnes PJ. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med. 2014;35(1):71-86.
doi pubmed - Daheshia M. Therapeutic inhibition of matrix metalloproteinases for the treatment of chronic obstructive pulmonary disease (COPD). Curr Med Res Opin. 2005;21(4):587-594.
doi pubmed - Salvi SS, Barnes PJ. Chronic obstructive pulmonary disease in non-smokers. Lancet. 2009;374(9691):733-743.
doi - Koksal H, Saygi A, Sariman N, Alici E, Yurtlu S, Yilmaz H, Duzgun Y. Evaluation of clinical and functional parameters in female subjects with biomass smoke exposure. Respir Care. 2013;58(3):424-430.
pubmed - Aaron SD, Angel JB, Lunau M, Wright K, Fex C, Le Saux N, Dales RE. Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;163(2):349-355.
doi pubmed - Halliwell B. The radical view: free radicals, health and disease. Odyssey. 1996,2(1):10-15.
- Maina JN, West JB. Thin and strong! The bioengineering dilemma in the structural and functional design of the blood-gas barrier. Physiol Rev. 2005;85(3):811-844.
doi pubmed - Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl J Med. 2011;365(6):537-547.
doi pubmed - Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr Opin Cell Biol. 2009;21(6):894-899.
doi pubmed pmc - Hansel TT, Barnes PJ. An atlas of chronic obstructive pulmonary disease: COPD. A resource for, teaching and lecturing. 1st ed. London: Taylor & Francis. 2003.
- Marwick JA, Adcock IM, Chung KF. Overcoming reduced glucocorticoid sensitivity in airway disease: molecular mechanisms and therapeutic approaches. Drugs. 2010;70(8):929-948.
doi pubmed - Tsiligianni IG, van der Molen T. A systematic review of the role of vitamin insufficiencies and supplementation in COPD. Respir Res. 2010;11:171.
doi pubmed pmc - Hwang JW, Rajendrasozhan S, Yao H, Chung S, Sundar IK, Huyck HL, Pryhuber GS, et al. FOXO3 deficiency leads to increased susceptibility to cigarette smoke-induced inflammation, airspace enlargement, and chronic obstructive pulmonary disease. J Immunol. 2011;187(2):987-998.
doi pubmed pmc - Kobayashi M, Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul. 2006;46:113-140.
- Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2008;8(3):183-192.
doi pubmed - Obeng EA, Boise LH. Caspase-12 and caspase-4 are not required for caspase-dependent endoplasmic reticulum stress-induced apoptosis. J Biol Chem. 2005;280(33):29578-29587.
doi pubmed - Hogg JC. Why does airway inflammation persist after the smoking stops? Thorax. 2006;61(2):96-97.
doi pubmed pmc - Shaban MM, Mohammed SA, Kam MA. Role de autoimmunity in the pathogenesis of chronic obstructive pulmonary disease. Egyp J Chest Tuberc. 2016;162(2):355-361.
- Shen TC, Wu BR, Chen HJ, Lin CL, Wei CC, Chen CH, Tu CY, et al. Risk of chronic obstructive pulmonary disease in female adult with Primary Sjogren Syndrome. Medicine. 2016;95(10):1-16.
doi pubmed pmc - Birring SS, Pavord ID. COPD: an autoimmune disease? Eur Respir J. 2011;38(2):484.
doi pubmed - Kirkham PA, Caramori G, Casolari P, Papi AA, Edwards M, Shamji B, Triantaphyllopoulos K, et al. Oxidative stress-induced antibodies to carbonyl-modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2011;184(7):796-802.
doi pubmed pmc - Rahman I, van Schadewijk AA, Crowther AJ, Hiemstra PS, Stolk J, MacNee W, De Boer WI. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;166(4):490-495.
doi pubmed - Kirkham PA, Spooner G, Ffoulkes-Jones C, Calvez R. Cigarette smoke triggers macrophage adhesion and activation: role of lipid peroxidation products and scavenger receptor. Free Radic Biol Med. 2003;35(7):697-710.
doi - Dalle-Donne I, Aldini G, Carini M, Colombo R, Rossi R, Milzani A. Protein carbonylation, cellular dysfunction, and disease progression. J Cell Mol Med. 2006;10(2):389-406.
doi pubmed pmc - Dalle-Donne I, Rossi R, Giustarini D, Milzani A, Colombo R. Protein carbonyl groups as biomarkers of oxidative stress. Clin Chim Acta. 2003;329(1-2):23-38.
doi - Eggleton P, Haigh R, Winyard PG. Consequence of neo-antigenicity of the ‘altered self’. Rheumatology (Oxford). 2008;47(5):567-571.
doi pubmed - Feghali-Bostwick CA, Gadgil AS, Otterbein LE, Pilewski JM, Stoner MW, Csizmadia E, Zhang Y, et al. Autoantibodies in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177(2):156-163.
doi pubmed pmc - Barreiro E, de la Puente B, Minguella J, Corominas JM, Serrano S, Hussain SN, Gea J. Oxidative stress and respiratory muscle dysfunction in severe chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;171(10):1116-1124.
doi pubmed - Allison ME, Fearon DT. Enhanced immunogenicity of aldehyde-bearing antigens: a possible link between innate and adaptive immunity. Eur J Immunol. 2000;30(10):2881-2887.
doi - Demedts IK, Bracke KR, Van Pottelberge G, Testelmans D, Verleden GM, Vermassen FE, Joos GF, et al. Accumulation of dendritic cells and increased CCL20 levels in the airways of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175(10):998-1005.
doi pubmed - Kang MJ, Homer RJ, Gallo A, Lee CG, Crothers KA, Cho SJ, Rochester C, et al. IL-18 is induced and IL-18 receptor alpha plays a critical role in the pathogenesis of cigarette smoke-induced pulmonary emphysema and inflammation. J Immunol. 2007;178(3):1948-1959.
doi pubmed - Di Stefano A, Caramori G, Gnemmi I, Contoli M, Vicari C, Capelli A, Magno F, et al. T helper type 17-related cytokine expression is increased in the bronchial mucosa of stable chronic obstructive pulmonary disease patients. Clin Exp Immunol. 2009;157(2):316-324.
doi pubmed pmc - Imaoka H, Hoshino T, Takei S, Kinoshita T, Okamoto M, Kawayama T, Kato S, et al. Interleukin-18 production and pulmonary function in COPD. Eur Respir J. 2008;31(2):287-297.
doi pubmed - Chen L, Chen G, Zhang MQ, Xiong XZ, Liu HJ, Xin JB, Zhang JC, et al. Imbalance between subsets of CD8(+) peripheral blood T cells in patients with chronic obstructive pulmonary disease. PeerJ. 2016;4:e2301.
doi pubmed pmc - Nurwidya F, Damayanti T, Yunus F. The role of innate and adaptive immune cells in the immunopathogenesis of chronic obstructive pulmonary disease. Tuberc Respir Dis (Seoul). 2016;79(1):5-13.
doi pubmed pmc - Garber K. Biochemistry: A radical treatment. Nature. 2012;489(7417):S4-6.
doi pubmed - Gregersen PK, Olsson LM. Recent advances in the genetics of autoimmune disease. Annu Rev Immunol. 2009;27:363-391.
doi pubmed pmc - Laurell CB, Eriksson S. The serum Alpha-L-Antitrypsin in families with Hypo-Alpha-L-Antitrypsinemia. Clin Chim Acta. 1965;11:395-398.
doi - Castaldi PJ, Cho MH, Cohn M, Langerman F, Moran S, Tarragona N, Moukhachen H, et al. The COPD genetic association compendium: a comprehensive online database of COPD genetic associations. Hum Mol Genet. 2010;19(3):526-534.
doi pubmed pmc - Greene CM, Hassan T, Molloy K, McElvaney NG. The role of proteases, endoplasmic reticulum stress and SERPINA1 heterozygosity in lung disease and alpha-1 anti-trypsin deficiency. Expert Rev Respir Med. 2011;5(3):395-411.
doi pubmed - Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med. 2008;359(22):2355-2365.
doi pubmed - Opitz B, van Laak V, Eitel J, Suttorp N. Innate immune recognition in infectious and noninfectious diseases of the lung. Am J Respir Crit Care Med. 2010;181(12):1294-1309.
doi pubmed - Alvarado A, Arce I. Immune recognition in lung diseases: basic research and clinical application. Clin Infect Immun. 2016;1(2):31-40.
doi - Natarajan V, Parinandi NL. Mitochondrial function in lung health and disease. New York, USA: Human Press; 2014.
doi - Cloonan SM, Choi AM. Mitochondria in lung disease. J Clin Invest. 2016;126(3):809-820.
doi pubmed pmc - Gray MW, Burger G, Lang BF. Mitochondrial evolution. Science. 1999;283(5407):1476-1481.
doi pubmed - Chinnery PF, Zeviani M. Mitochondrial matchmaking. N Engl J Med. 2016;375(19):1894-1896.
doi pubmed - Scheffler IE. Mitochondria. 2nd ed. Hoboken: Wiley-Liss; 2007.
doi - Huttemann M, Lee I, Gao X, Pecina P, Pecinova A, Liu J, Aras S, et al. Cytochrome c oxidase subunit 4 isoform 2-knockout mice show reduced enzyme activity, airway hyporeactivity, and lung pathology. FASEB J. 2012;26(9):3916-3930.
doi pubmed pmc - Bialas AJ, Sitarek P, Milkowska-Dymanowska J, Protrowski WJ, Gorski P . The role of mitochondrial and oxidative/antioxidative imbalance in pathobiology of chronic obstructive pulmonary disease. Oxid Med Cel Longev. 2016;2016:7808576.
- Kang MJ, Shadel GS. A mitochondrial perspective of chronic obstructive pulmonary disease pathogenesis. Tuberc Respir Dis (Seoul). 2016;79(4):207-213.
doi pubmed pmc - Ristow M, Schmeisser K. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose Response. 2014;12(2):288-341.
doi pubmed pmc - Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem. 2010;47:69-84.
doi pubmed pmc - Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11(12):872-884.
doi pubmed - Bhatia-Kissova I, Camougrand N. Mitophagy is not induced by mitochondrial damage but plays a role in the regulation of cellular autophagic activity. Autophagy. 2013;9(11):1897-1899.
doi pubmed - Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122(5):669-682.
doi pubmed - Arnoult D, Soares F, Tattoli I, Girardin SE. Mitochondria in innate immunity. EMBO Rep. 2011;12(9):901-910.
doi pubmed pmc - Vlahos R, Bozinovski S. Role of alveolar macrophages in chronic obstructive pulmonary disease. Front Immunol. 2014;5:435.
doi pubmed pmc - Byrne AJ, Mathie SA, Gregory LG, Lloyd CM. Pulmonary macrophages: key players in the innate defence of the airways. Thorax. 2015;70(12):1189-1196.
doi pubmed - Falk MJ, Decherney A, Kahn JP. Mitochondrial replacement techniques - implications for the clinical community. N Engl J Med. 2016;374(12):1103-1106.
doi pubmed pmc - Phimister EG. Mitochondral donation clearing the final regulatory hundle in the United Kingdom. N Engl J Med. 2017;376(2):171-173.
doi pubmed - Picard M, Wallace DC, Burelle Y. The rise of mitochondria in medicine. Mitochondrion. 2016;30:105-116.
doi pubmed pmc - Ribera F, N’Guessan B, Zoll J, Fortin D, Serrurier B, Mettauer B, Bigard X, et al. Mitochondrial electron transport chain function is enhanced in inspiratory muscles of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2003;167(6):873-879.
doi pubmed - Rabinovich RA, Vilaro J. Structural and functional changes of peripheral muscles in COPD patients. Curr Opin Pulm Med. 2010;162(2):123-133.
doi pubmed pmc - Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014;124(9):3987-4003.
doi pubmed pmc - Lommatzsch M, Cicko S, Muller T, Lucattelli M, Bratke K, Stoll P, Grimm M, et al. Extracellular adenosine triphosphate and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181(9):928-934.
doi pubmed - van der Toorn M, Slebos DJ, de Bruin HG, Leuvenink HG, Bakker SJ, Gans RO, Koeter GH, et al. Cigarette smoke-induced blockade of the mitochondrial respiratory chain switches lung epithelial cell apoptosis into necrosis. Am J Physiol Lung Cell Mol Physiol. 2007;292(5):L1211-1218.
doi pubmed - Tashkin DP, Celli B, Senn S, Burkhart D, Kesten S, Menjoge S, Decramer M, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med. 2008;359(15):1543-1554.
doi pubmed - Magnussen H, Disse B, Rodriguez-Roisin R, Kirsten A, Watz H, Tetzlaff K, Towse L, et al. Withdrawal of inhaled glucocorticoids and exacerbations of COPD. N Engl J Med. 2014;371(14):1285-1294.
doi pubmed - Dekhuijzen PN, Van Beurden WJ. The role for N-acetyl cysteine in the management of COPD. Int J Chron Obstruct Pulmon Dis. 2006;1(2):99-106.
doi pubmed pmc - Decramer M, Rutten-van Molken M, Dekhuijzen PN, Troosters T, van Herwaarden C, Pellegrino R, van Schayck CP, et al. Effects of N-acetylcysteine on outcomes in chronic obstructive pulmonary disease (Bronchitis Randomized on NAC Cost-Utility Study, BRONCUS): a randomised placebo-controlled trial. Lancet. 2005;365(9470):1552-1560.
doi - Tse HN, Raiteri L, Wong KY, Yee KS, Ng LY, Wai KY, Loo CK, et al. High-dose N-acetylcysteine in stable COPD: the 1-year, double-blind, randomized, placebo-controlled HIACE study. Chest. 2013;144(1):106-118.
doi pubmed - Stav D, Raz M. Effect of N-acetylcysteine on air trapping in COPD: a randomized placebo-controlled study. Chest. 2009;136(2):381-386.
doi pubmed - Tse HN, Tseng CZ. Update on the pathological processes, molecular biology, and clinical utility of N-acetylcysteine in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2014;9:825-836.
doi pubmed pmc - Rahman I, MacNee W. Antioxidant pharmacological therapies for COPD. Curr Opin Pharmacol. 2012;12(3):256-265.
doi pubmed pmc - Pareek TK, Belkadi A, Kesavapany S, Zaremba A, Loh SL, Bai L, Cohen ML, et al. Triterpenoid modulation of IL-17 and Nrf-2 expression ameliorates neuroinflammation and promotes remyelination in autoimmune encephalomyelitis. Sci Rep. 2011;1:201.
doi pubmed pmc - Albert RK. "Lies, damned lies …" and observational studies in comparative effectiveness research. Am J Respir Crit Care Med. 2013;187(11):1173-1177.
doi pubmed - Boueiz A, Damarla M, Hassoun PM. Xanthine oxidoreductase in respiratory and cardiovascular disorders. Am J Physiol Lung Cell Mol Physiol. 2008;294(5):L830-840.
doi pubmed - Jaquet V, Marcoux J, Forest E, Leidal KG, McCormick S, Westermaier Y, Perozzo R, et al. NADPH oxidase (NOX) isoforms are inhibited by celastrol with a dual mode of action. Br J Pharmacol. 2011;164(2b):507-520.
doi pubmed pmc - Churg A, Marshall CV, Sin DD, Bolton S, Zhou S, Thain K, Cadogan EB, et al. Late intervention with a myeloperoxidase inhibitor stops progression of experimental chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185(1):34-43.
doi pubmed - Yao H, Arunachalam G, Hwang JW, Chung S, Sundar IK, Kinnula VL, Crapo JD, et al. Extracellular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc Natl Acad Sci U S A. 2010;107(35):15571-15576.
doi pubmed pmc - Seimetz M, Parajuli N, Pichl A, Veit F, Kwapiszewska G, Weisel FC, Milger K, et al. Inducible NOS inhibition reverses tobacco-smoke-induced emphysema and pulmonary hypertension in mice. Cell. 2011;147(2):293-305.
doi pubmed - Sato A, Hoshino Y, Hara T, Muro S, Nakamura H, Mishima M, Yodoi J. Thioredoxin-1 ameliorates cigarette smoke-induced lung inflammation and emphysema in mice. J Pharmacol Exp Ther. 2008;325(2):380-388.
doi pubmed - Couillin I, Vasseur V, Charron S, Gasse P, Tavernier M, Guillet J, Lagente V, et al. IL-1R1/MyD88 signaling is critical for elastase-induced lung inflammation and emphysema. J Immunol. 2009;183(12):8195-8202.
doi pubmed - Dima E, Koltsida O, Katsaounou P, Vakali S, Koutsoukou A, Koulouris NG, Rovina N. Implication of Interleukin (IL)-18 in the pathogenesis of chronic obstructive pulmonary disease (COPD). Cytokine. 2015;74(2):313-317.
doi pubmed - Kurien BT, Scofield RH. Autoimmunity and oxidatively modified autoantigens. Autoimmun Rev. 2008;7(7):567-573.
doi pubmed pmc - Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol. 2014;15(10):634-646.
doi pubmed pmc
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cellular and Molecular Medicine Research is published by Elmer Press Inc.